Pipeline nf-core/ampliseq
Introduzione
La pipeline nf-core/ampliseq proviene dalla community nf-core, dedita alla costruzione e condivisione di pipeline bioinformatiche gestite con Nextflow. Essa effettua il sequenziamento ed il denoising degli ampliconi; può disporre, inoltre, di diversi database per l'assegnazione tassonomica (tra cui database per sequenze 16S, ITS, CO1 e 18S). La pipeline supporta dati provenienti da dispositivi Illumina, PacBio and IonTorrent.
La versione della pipeline nf-core/ampliseq disponibile in GenPat è la 2.7.1.
Pagina ufficiale della versione 2.7.1 di nf-core/ampliseq sul sito della community "nf-core": https://nf-co.re/ampliseq/2.7.1.
Pagina GitHub di nf-core/ampliseq: https://github.com/nf-core/ampliseq.

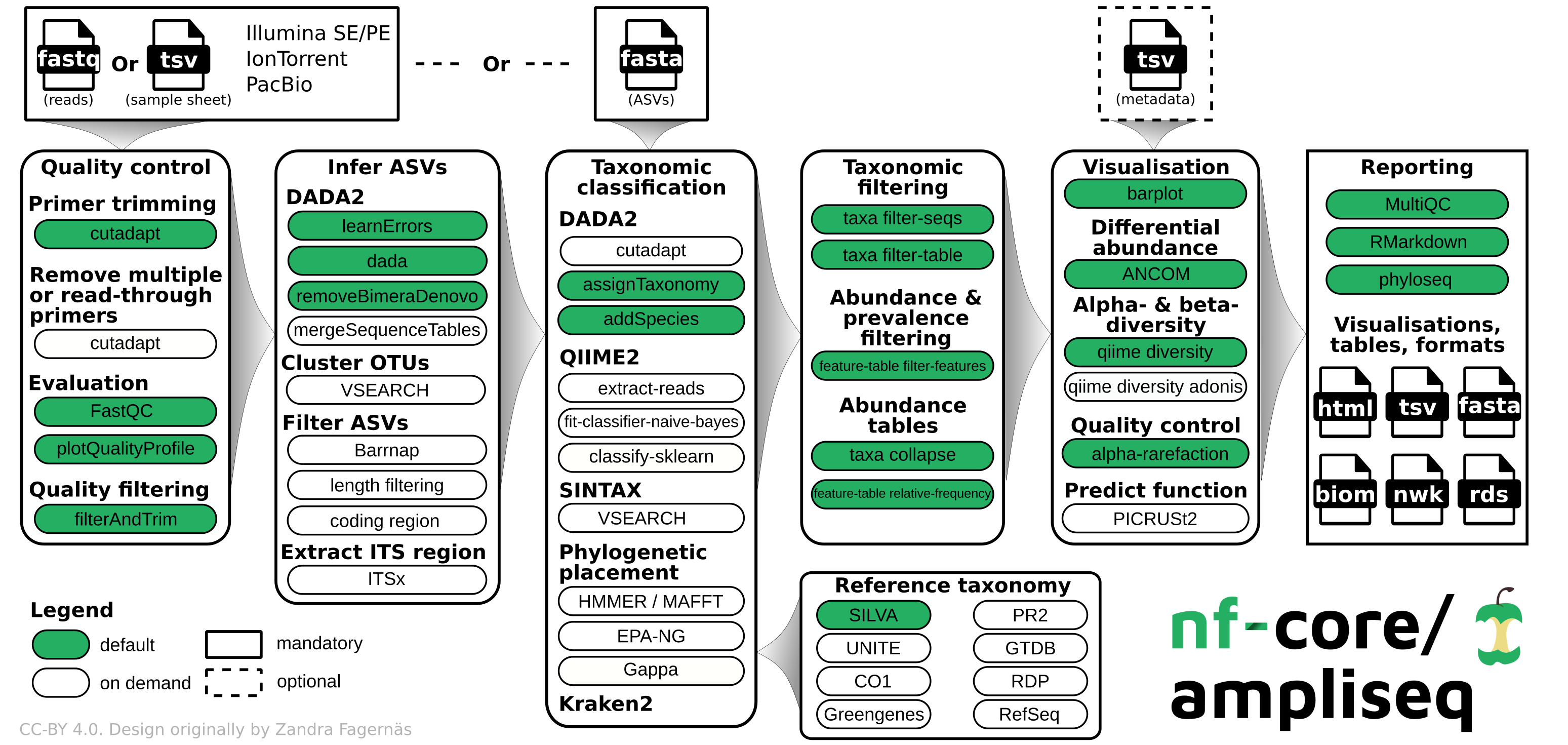 Immagine di proprietà di ©nf-core: https://nf-co.re/ampliseq/2.7.1.
Immagine di proprietà di ©nf-core: https://nf-co.re/ampliseq/2.7.1.
Le analisi di default di nf-core/ampliseq (in verde nell'immagine sovrastante) sono quelle che vengono eseguite in GenPat. Alcune delle analisi "on demand" possono essere aggiunte sfruttando i parametri addizionali nel lancia analisi; alcune analisi di default, inoltre, possono essere saltate, sempre sfruttando i parametri addizionali. Si invita a consultare le sezioni "parameters" e "parameters: skipping specific steps" della guida ufficiale di nf-core/ampliseq per sfruttare al meglio i parametri addizionali ed il campo di testo per i parametri shell-like.
Lancia la pipeline nf-core/ampliseq
Nel sistema di lancio analisi, è possibile usare il filtro in alto, per visualizzare esclusivamente le pipelines. Una volta selezionata la pipeline nf-core/ampliseq, nella pagina dedicata al lancio di analisi, il sistema passerà ad un'interfaccia di conferma.
Le fasi della pipeline sono riassunte nello schema all'inizio della pagina.
La pipeline supporta file di sequenze provenienti da dispositivi di sequenziamento Illumina, IonTorrent o PacBio:
L'interfaccia per la selezione dell'input mette a disposizione la modalità di selezione input avanzata, per permettere l'utilizzo di input processati da metodi diversi, usati a monte nel flusso di analisi.
Parametri addizionali
Di seguito, sono elencati i parametri addizionali disponibili nella sezione apposita del sistema di lancio:
FW_primereRV_primer: campi di testo nei quali incollare la sequenza, rispettivamente, dei primer forward e dei primer reverse; si tratta di un'operazione necessaria ai fini del trimming dei primers;sample_inference, modalità di inferenza per il calcolo delle ASV:independent: modalità "risparmio", più veloce e leggera ma con una ridotta sensibilità;pooled: modalità "performance", con maggiore sensibilità ma più pesante da eseguire;pseudo: "pseudo-pooled". Modalità bilanciata;
trunclenf,trunclenretrunc_qmin: la fase di denoising richiede read della stessa lunghezza. I parametri per il troncamento sono campi di testo in cui specificare manualmente la posizione nucleotidica da usare come valore di cutoff per la forward read (trunclenf) e per la reverse read (trunclenr). Le sequenze più lunghe vengono troncate laddove quelle più corte vengono scartate.trunc_qminpermette di specificare un valore minimo di qualità della chiamata del nucleotide: setrunclenfetrunclenrnon vengono specificati, i valori di cutoff vengono determinati automaticamente, troncando quando il valore mediano del quality score scende sotto la soglia specificata.dada_ref_db: selezione del database usato da DADA2 per l'assegnazione tassonomica (in GenPat sono attualmente disponibili il database di rRNA 12S "mito-all" e di rRNA 16S "silva").Reads: selezione della tipologia di read (paired-end/single-end).Custom metadata (from IZSBOX): permette di indicare un file di metadati presente nel proprio IZSbox (incollare il percorso del file).Custom parameters: gli altri parametri di nf-core/ampliseq sono disponibili come modificatori, da scrivere nel campo di testo come comandi da terminale. La guida ufficiale contiene tutte le informazioni necessarie.
Si invita a consultare le descrizioni dei parametri direttamente nella guida ufficiale nf-core/ampliseq: https://nf-co.re/ampliseq/2.7.1/parameters.
Una volta lanciata la pipeline, la pagina genererà un link alla sezione Controllo analisi, per permettere di visualizzare lo stato del processo. L'utente verrà notificato dal sistema sia una volta lanciata con successo la pipeline, sia al termine dell'esecuzione.
Cartella dei risultati
La scheda riassuntiva della pipeline completata con successo permette di accedere alla cartella dei risultati, in modo da recuperare i files tabulari e le immagini dei grafici; essa fornisce, inoltre, link diretti per la visualizzazione delle immagini e dei report.
La cartella dei risultati, Result folder, è accessibile cliccando sul link presente all'interno della scheda dell'analisi, nella sezione Dati risultato. All'interno della conseguente cartella results, è possibile trovare i files prodotti dalla pipeline nf-core/ampliseq.
La tabella in basso presenta la lista delle principali cartelle ed il loro contenuto, insieme ad alcune informazioni utili.
| Cartella | Contenuto |
|---|---|
results/ |
sotto-cartelle dei software e tabella riassuntiva (overall_summary.tsv) |
results/cutadapt |
log di cutadapt (DSXXXXXXXX.trimmed.cutadapt.log) e tabella riassuntiva (cutadapt_summary.tsv) |
results/dada2 |
tabelle .tsv delle statistiche di DADA2, degli ASV trovati e fasta delle sequenze degli ASV (ASV_seqs.fasta) |
results/dada2/QC |
grafici del controllo qualità in formato .pdf |
results/input |
file dei metadati (metadata.tsv) |
results/multiqc |
report di multiQC in formato .html, cartelle dei dati di output e dei grafici di MultiQC |
results/qiime |
cartelle dei dati prodotti da QIIME: abundance_tables, alpha-rarefaction, barplot, diversity, phylogenetic_tree, rel_abundance_tables, representative_sequences |
results/summary_report |
grafici in formato immagine .svg e report in formato .html |