1PP_downsampling
Introduzione
Il downsampling è definibile come un processo di riduzione di read depth (coverage verticale), in specifiche posizioni o all'interno di determinate regioni del genoma, allo scopo di diminuire il numero di read non informative ed aumentare la qualità dell'assembly e delle analisi a valle. BBnorm normalizza la read depth usando un hashing di k-mers.
I protocolli di sequenziamento possono spesso provocare l'accumulo di read per una sezione del genoma. L'eccesso di informazione, in questi casi, rallenta l'esecuzione delle analisi a valle, costrette a processare un volume maggiore di dati, pur non essendo tutti questi informativi. In questi casi, il downsampling permette di evitare l'allungamento dei tempi di calcolo, scartando coppie di read in eccesso, fino al raggiungimento di un valore limite specificato, ovvero il coverage verticale desiderato.

Lancia Analisi 1PP_downsampling
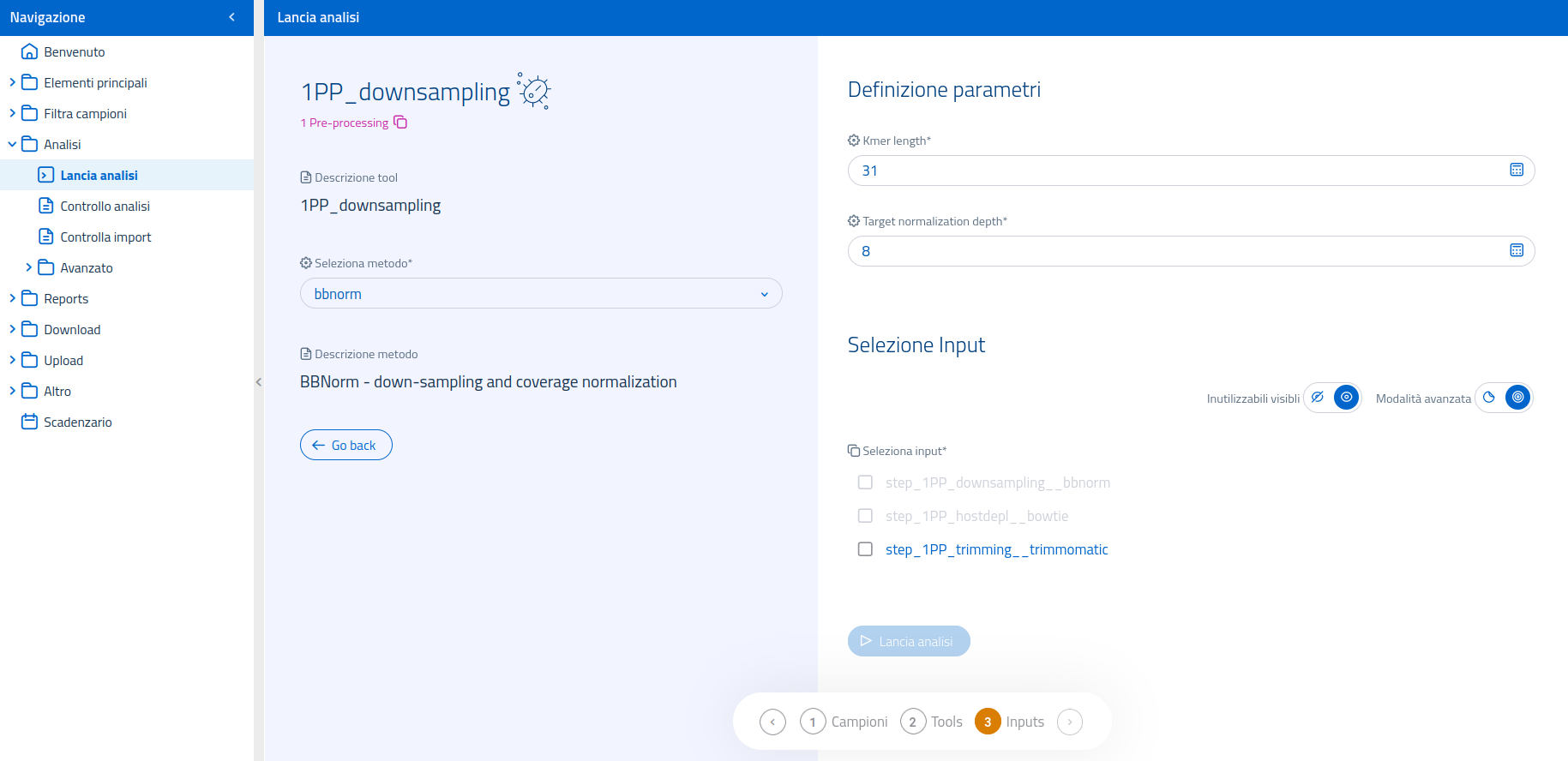
Una volta selezionata l'analisi 1PP_downsampling nella pagina dedicata al lancio di analisi, sarà possibile selezionare il software bioinformatico ("metodo") da usare tra quelli disponibili per l'analisi. Il tool utilizzabile, in questo caso, è:
- BBnorm - strumento di correzione degli errori e normalizzazione basato su K-mer.
Manuale di BBnorm: https://manpages.debian.org/unstable/bbmap/bbnorm.sh.1.en.html
Nella sezione dedicata alla selezione dei parametri, sarà necessario indicare sia le read di input sia alcuni parametri addizionali, atti a determinare il coverage verticale finale che si desidera avere per i genomi dei campioni.
La sezione di selezione degli input mette a disposizione la modalità di selezione input avanzata. E' infatti possibile che il downsampling venga lanciato a seguito di analisi eseguite a monte e diverse tra loro:
- step_1PP_trimming
- step_1PP_hostdepl
- step_1PP_downsampling
Una volta lanciata l'analisi, la pagina genererà un link alla sezione Controllo analisi, per permettere di visualizzare lo stato del processo. L'utente verrà notificato dal sistema sia una volta lanciata con successo l'analisi, sia al termine dell'esecuzione.
Parametri
Una panoramica dei parametri di BBnorm è disponibile al seguente link: https://manpages.debian.org/unstable/bbmap/bbnorm.sh.1.en.html.
I 2 parametri "kmer length" e "Target" dipendono da:
- specie del campione (e quindi lunghezza del genoma);
- per "Target", dalla lunghezza dei kmer da usare, come specificata nel parametro "kmer length".
Kmer length ha un valore di default di 31. Il parametro supporta una qualunque lunghezza per i kmer ma viene consigliata una lunghezza inferiore a 32 per una maggiore efficienza del tool.
Target indica la normalizzazione dei k-mer usati da BBnorm per eseguire il downsampling.
Attenzione: il parametro "Target normalization value" non corrisponde al coverage verticale finale che si otterrà con l'esecuzione del tool; piuttosto, secondo la guida ai parametri di BBnorm, esso controlla la profondità verticale dei k-mers che vengono usati dal tool per normalizzare le read.
Per via del funzionamento del software è pertanto necessario determinare sperimentalmente il valore del secondo parametro, in relazione al primo ("kmer length") e alla specie.
Di seguito sono riportati due esempi per Listeria monocytogenes:
- specie L. monocytogenes, kmer length = 30, target = 31 -> vertical coverage = 40X
- specie L. monocytogenes, kmer length = 30, target = 8 -> vertical coverage = 10X
Cartella dei risultati
Per consultare la guida sul download dei files dalla piattaforma si faccia riferimento all'apposita pagina.
La cartella dei risultati, Result folder, è accessibile cliccando sul link presente all'interno della scheda dell'analisi, nella sezione Dati risultato. All'interno della conseguente cartella results, è possibile trovare 2 sotto-cartelle:
- meta: (metadati) in cui vengono salvati i file di log e di configurazione del processo eseguito.
- result: in cui sono salvati i file con i risultati prodotti dall'analisi.
La tabella in basso presenta la lista di files presenti nelle cartelle, insieme ad alcune informazioni utili.
| File | Descrizione | Posizione |
|---|---|---|
| DSXXXXXXXX-DTXXXXXX_ID_bbnorm_kXX_tX_R1.fastq.gz | read 1 (R1) downsampled | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_bbnorm_kXX_tX_R2.fastq.gz | read 2 (R2) downsampled | cartella "result" |