WGSBAC Pipeline - IAEA (Brucella)
Introduzione
La pipeline Wgsbac effettua la genotipizzazione e la caratterizzazione di isolati batterici utilizzando, come input, i file fastq.gz provenienti dal sequenziamento del genoma completo (whole-genome-sequencing, WGS). Nella piattaforma, sono implementati i workflow per campioni di Brucella. Per un elenco completo delle analisi disponibili nel workflow, è possibile consultare la pagina GitLab di WGSBAC.
- Pagina ufficiale GitLab di WGSBAC: https://gitlab.com/FLI_Bioinfo/WGSBAC
- Autori: Jörg Linde (@joerg.linde) e Mostafa Abdel-Glil (@Mostafa.Abdel-Glil).

Lanciare la pipeline Wgsbac - IAEA
Altri microorganismi, che possono essere analizzati con la pipeline originale, non sono supportati dall'installazione nella presente piattaforma.
La pipeline WGSBAC esegue una gamma di tool bioinformatici suddivisibili in:
- controllo qualità (QC) delle read e trimming;
- ricostruzione della sequenza;
- QC dell'assemblaggio e controllo di contaminazione;
- annotazione del genoma;
- tipizzazione;
- clustering gerarchico.
Una lista dettagliata di tutti i software eseguibili dalla pipeline è presente nella pagina ufficiale GitLab di WGSBAC.
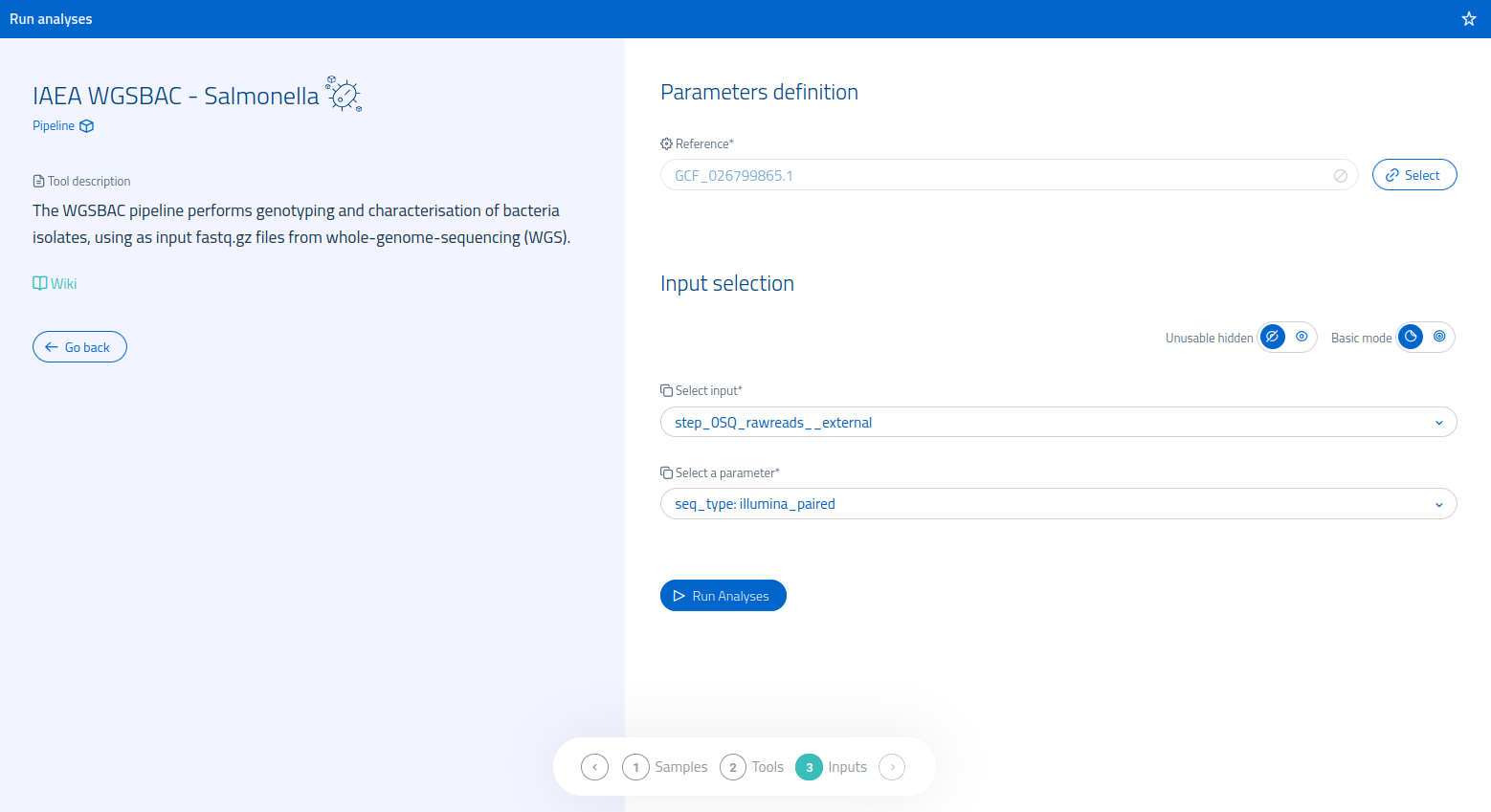
Dopo aver selezionato la versione WGSBAC per il microorganismo di interesse, un'interfaccia di conferma permetterà di proseguire con la selezione dei parametri dell'analisi.
Gli input necessari sono reads fastq compresse (.fastq.gz), che possono essere fornite come:
- step_0SQ_rawreads
- step_1PP_trimming
Si noti che l'opzione step_1PP_trimming, come input, viene mantenuta solo per comodità dell'utente poiché la pipeline WGSBAC effettua, di default, il trimming con fastp.
Per poter eseguire alcune delle analisi della pipeline, inoltre, è necessario specificare un genoma reference per il microorganismo selezionato.
Nota importante: si ricordi che la pipeline WGSBAC può essere utilizzata solo su read Illumina paired end. Non sono attualmente supportate altre tecnologie di sequenziamento.
Una volta lanciata la pipeline, la pagina genererà un link alla sezione Controllo analisi, per permettere di visualizzare lo stato del processo. L'utente verrà notificato dal sistema sia una volta lanciata con successo la pipeline, sia al termine dell'esecuzione.
Cartella dei risultati
I file dei risultati della pipeline WGSBAC includono gli outputs di ogni singola analisi. In basso, sono elencate le cartelle dei risultati di ciascuna analisi. Per una lista completa si rimanda alla pagina GitLab ufficiale WGSBAC: (https://gitlab.com/FLI_Bioinfo/WGSBAC).
La cartella dei risultati, Result folder, è accessibile cliccando sul link presente all'interno della scheda dell'analisi, nella sezione Dati risultato. All'interno della conseguente cartella results, è possibile trovare i risultati delle singole analisi.
- Per l'esecuzione su campioni di Brucella:
| Analisi - Tool/DB | Cartella |
|---|---|
| controllo di contaminazione - confindr | wgsbac_results > confindr |
| calcolo del coverage | wgsbac_results > coverage |
| trimming e QC - fastp | wgsbac_results > fastp |
| file fastq decompressi | wgsbac_results > fastq |
| QC delle raw reads - fastqc | wgsbac_results > fastqc |
| fasta del reference | wgsbac_results > finalAssembly |
| file fastq.gz di input | wgsbac_results > input |
| controllo di contaminazione - kraken2 | wgsbac_results > kraken |
| Multi Lucus Sequence Typing - mlst | wgsbac_results > mlst |
| QC - multiQC | wgsbac_results > multiqc |
| QC - fastQC | wgsbac_results > multiqc_fastqc |
| identificazione dei plasmidi - plasmidfinder/platon | wgsbac_results > plasmids |
| QC dell'assemblaggio - Quast | wgsbac_results > quast |
| assemblaggio de novo - Shovill | wgsbac_results > rawassembly_res |
| chiamata delle varianti - Snippy | wgsbac_results > snippy |
| identificazione della specie - Sourmash | wgsbac_results > sourmash |