1PP_trimming
Introduzione
Il trimming ha lo scopo di rimuovere i residui di bassa qualità dalle read ottenute dal sequenziatore.
I tool disponibili per il trimming in GenPat sono "trimmomatic" e "fastp". L'analisi 1PP_trimming è costituita dall'esecuzione di uno dei tools per trimming e di fastQC. Quest'ultimo elabora le statistiche di qualità delle reads e viene eseguito sia sulle raw reads, prima del trimming, sia sulle nuove reads trimmate.

Lancia 1PP_trimming
Una volta selezionata l'analisi 1PP_trimming nella pagina dedicata al lancio di analisi, sarà possibile selezionare il software bioinformatico da usare tra quelli disponibili per l'analisi. I tools utilizzabili, in questo caso, sono:
-
trimmomatic - tool di trimming delle read per dati NGS Illumina
Pagina GitHub di trimmomatic: https://github.com/usadellab/Trimmomatic
-
fastp - pre-processamento all-in-one per i file fastQ
Pagina GitHub di fastp: https://github.com/OpenGene/fastp
-
chopper - filtraggio e trimming dei file fastQ provenienti da sequenziamento a long reads, come PacBio o ONT
Pagina GitHub di chopper: https://github.com/wdecoster/chopper
Nota 1: Per il trimming di long reads usare esclusivamente chopper; per il trimming di short reads usare trimmomatic o fastp.
Nota 2: La guida ai parametri di Chopper è disponibile nella sua pagina GitHub ufficiale.
- seqkit - un kit di tool multipiattaforma e ultrarapido per la manipolazione di file fastA/fastQ
pagina GitHub di seqkit: https://bioinf.shenwei.me/seqkit/usage/#grep
Nota 3: Questo strumento elimina, dalle reads, tutte le letture in cui è presente la sequenza dell'adattatore indicata in "Custom adapter sequence", utilizzando la funzione "grep" del tool.
Nota 4: Solo per illumina reads. Per questa analisi è possibile utilizzare come input sia l'analisi 0SQ_rawreads che 1PP_trimming.
Nell'ultima sezione viene invece specificato l'input: poichè il trimming viene eseguito sui fastQ ottenuti dal sequenziatore, le scelte disponibili saranno "step_0SQ_rawreads__fastq" per i fastq delle reads interne (codice 20XX.TE.XXXX.X.X) e "step_0SQ_rawreads__external", per i fastq importati (codice 20XX.EXT.XXXX.X.X). La schermata di selezione input di 1PP_trimming mette a disposizione dell'utente la modalità di selezione input avanzata, in quanto l'analisi è eseguibile sia su file di origine interna, sia su file importati.
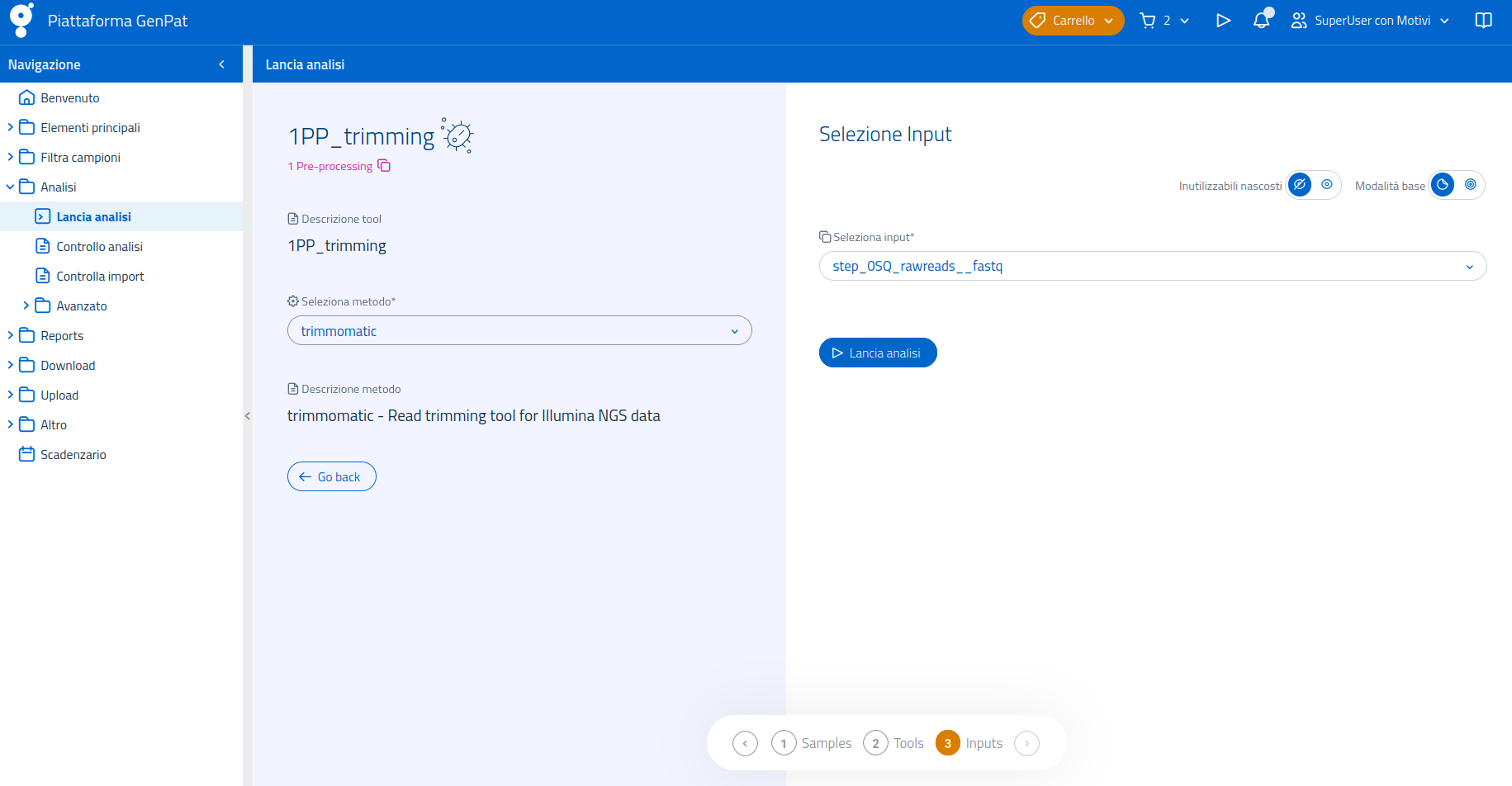
Una volta lanciata l'analisi, la pagina genererà un link alla sezione Controllo analisi, per permettere di visualizzare lo stato del processo. L'utente verrà notificato dal sistema sia una volta lanciata con successo l'analisi, sia al termine dell'esecuzione.
Cartella dei risultati
Per consultare la guida sul download dei files dalla piattaforma si faccia riferimento all'apposita pagina.
La cartella dei risultati, Result folder, è accessibile cliccando sul link presente all'interno della scheda dell'analisi, nella sezione Dati risultato. All'interno della conseguente cartella results, è possibile trovare 3 sotto-cartelle:
- meta: (metadati) in cui vengono salvati i file di log e di configurazione del processo eseguito;
- qc: (quality check) in cui si trovano i file del controllo qualità. In questa cartella sono presenti le cartelle meta e result. Per questa analisi, il controllo qualità viene eseguito con fastqc. Il calcolo del QC non viene eseguito in caso di lancio analisi con Seqkit;
- result: in cui sono salvati i file con i risultati prodotti dall'analisi.
Le tabelle in basso presentano le liste di file presenti nelle cartelle, in base al tool usato, insieme ad alcune informazioni utili. Si noti che nel nome del file verrà riportato il nome del programma che è stato scelto per l'analisi.
Trimmomatic
| File | Descrizione | Posizione |
|---|---|---|
| DSXXXXXXXX-DTXXXXXX_ID_R1_trimmomatic.fastq | read 1 (R1) trimmata | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_R2_trimmomatic.fastq | read 2 (R2) trimmata | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_unpaired_trimmomatic.fastq | unpaired reads trimmate | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_R1_trimmomatic_fastqc.html | qualità delle read R1 | cartella qc > result |
| DSXXXXXXXX-DTXXXXXX_ID_R1_trimmomatic_fastqc.zip | qualità R1 (file zip) | cartella qc > result |
| DSXXXXXXXX-DTXXXXXX_ID_R2_trimmomatic_fastqc.html | qualità delle read R2 | cartella qc > result |
| DSXXXXXXXX-DTXXXXXX_ID_R2_trimmomatic_fastqc.zip | qualità R2 (file zip) | cartella qc > result |
| DSXXXXXXXX-DTXXXXXX_ID_unpaired_trimmomatic_fastqc.html | qualità delle unpaired read | cartella qc > result |
| DSXXXXXXXX-DTXXXXXX_ID_unpaired_trimmomatic_fastqc.zip | qualità delle unpaired read (file zip) | cartella qc > result |
fastp
| File | Descrizione | Posizione |
|---|---|---|
| DSXXXXXXXX-DTXXXXXX_ID_fastp_R1.fastq.gz | read 1 (R1) trimmata | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_fastp_R2.fastq.gz | read 2 (R2) trimmata | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_fastp_unpaired.fastq.gz | unpaired read trimmate | cartella "result" |
chopper
| File | Descrizione | Posizione |
|---|---|---|
| filtered_reads.fastq.gz | long read trimmate | cartella "result" |