2AS_mapping
Introduzione
L'analisi 2AS_mapping esegue l'assembly con reference (o mapping) delle sequenze.

Lancia analisi 2AS_mapping
Una volta selezionata l'analisi 2AS_mapping nella pagina dedicata al lancio di analisi, sarà possibile selezionare il tool bioinformatico ("metodo") da usare tra quelli disponibili per l'analisi. I tool utilizzabili per questa analisi sono:
-
bowtie2 - uno strumento ultrarapido ed efficiente in termini di memoria per l'allineamento delle read di sequenziamento a lunghe sequenze reference.
Pagina GitHub di bowtie2: https://github.com/BenLangmead/bowtie2
-
iVar - pacchetto informatico per il sequenziamento virale basato su ampliconi.
Pagina GitHub di iVar: https://github.com/andersen-lab/ivar
-
Snippy - chiamata rapida delle varianti aploidi e allineamento del genoma core.
Pagina GitHub di Snippy: https://github.com/tseemann/snippy
Nota: Nel caso si utilizzi il metodo snippy, è indispensabile che il campione scelto abbia l'analisi 4AN_genes associata (Prokka).
- medaka - tool per creare sequenze "consensus" e chiamate di varianti, a partire da dati di sequenziamento Nanopore.
Pagina GitHub di medaka: https://github.com/nanoporetech/medaka
Nota: medaka è un tool open source sviluppato dalla Oxford Nanopore Technologies Ltd., da usare esclusivamente su dati di sequenziamento provenienti da apparati Nanopore.
Nella sezione dedicata alla definizione dei parametri, sarà necessario selezionare un reference per il mapping, dalla tabella pop-up accessibile cliccando sul tasto Seleziona reference. Il pop-up elenca sequenze di due tipologie, entrambe utilizzabili come reference:
- reference fasta files: file importati da database esterni - come ad es. NCBI - (elencati nella tabella delle reference di GenPat: https://genpat.izs.it/cmdbuild/ui/#classes/T_REFERENCE/cards);
- la consensus o il de novo assembly di un campione presente in GenPat (database interni).
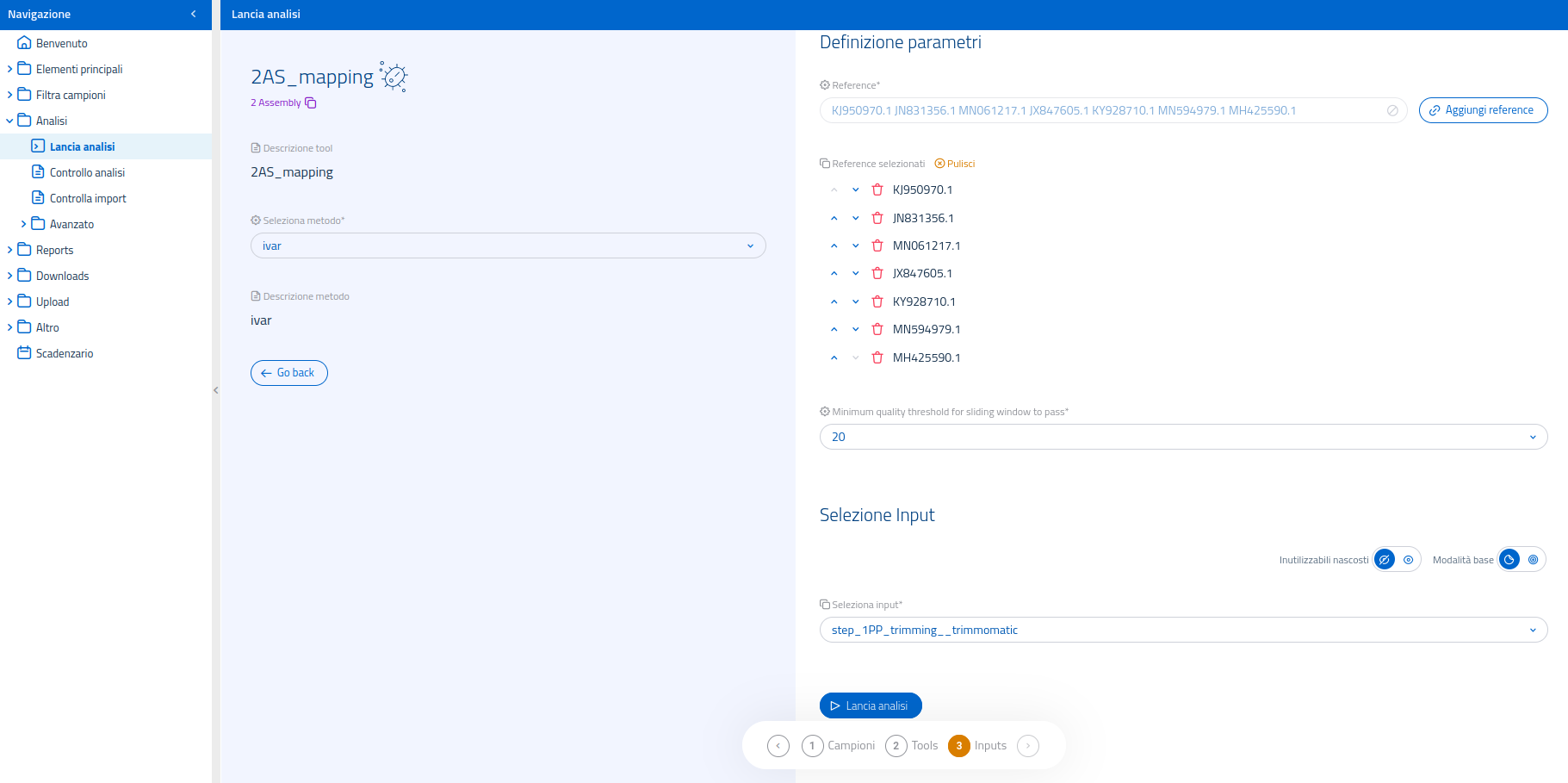
Con il metodo "ivar", è possibile selezionare più di un reference (si faccia riferimento alla sezione "Reference multipli", più in basso).
L'interfaccia per la selezione dell'input mette a disposizione la modalità di selezione input avanzata, per permettere l'utilizzo di input processati da metodi diversi, usati a monte nel flusso di analisi.
I possibili input sono gli stessi utilizzabili anche per 2AS_denovo:
- step_1PP_trimming
- step_1PP_hostdepl
- step_1PP_downsampling
- step_1PP_generated
- step_3TX_class
- step_4AN_AMR
Una volta lanciata l'analisi, la pagina genererà un link alla sezione Controllo analisi, per permettere di visualizzare lo stato del processo. L'utente verrà notificato dal sistema sia una volta lanciata con successo l'analisi, sia al termine dell'esecuzione.
Reference multipli
Se 2AS_mapping viene lanciato con Ivar, è possibile selezionare più di un reference, come mostrato nell'immagine riportata di seguito. La guida all'uso del sistema di selezione di reference multipli è consultabile nell'apposita sezione della Wiki sul lancio di analisi.
Cartella dei risultati
Per consultare la guida sul download dei files dalla piattaforma si faccia riferimento all'apposita pagina.
La cartella dei risultati, Result folder, è accessibile cliccando sul link presente all'interno della scheda dell'analisi, nella sezione Dati risultato. All'interno della conseguente cartella results, è possibile trovare 2 sotto-cartelle:
- meta: (metadati) in cui vengono salvati i file di log e di configurazione del processo eseguito;
- result: in cui sono salvati i files con i risultati prodotti dall'analisi.
Le tabelle sottostanti elencano i files prodotti dai tool disponibili per 2AS_mapping.
Bowtie2
| File | Descrizione | Posizione |
|---|---|---|
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID.fasta | file della consensus | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID.sorted.bam | file di allineamento in formato .bam (Binary Alignment Map) | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID.sorted.bam.bai | file .bai (indice del file BAM) | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID.var.flt.vcf | file delle varianti identificate in formato .vcf | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID_coverage_plot.png | grafico della distribuzione del coverage | cartella "result" |
iVar e Snippy
Si noti che l'uso di ivar comprende l'esecuzione dei tool snippy, samtools e, infine, ivar, mentre 2AS_mapping__snippy esegue esclusivamente il software da cui prende il nome.
| File | Descrizione | Posizione |
|---|---|---|
| DSXXXXXXXX-DTXXXXXX_ID_ivar_REFID.fasta | consensus ottenuta con ivar (Nota: 2AS_mapping__snippy non produce questo file) | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.aligned.fa | versione del reference con - nelle posizioni con sequencing depth = 0 (numero di read che allineano in quella posizione = 0) e N per depth compresa tra 0 ed il numero minimo di reads considerate nel coverage dei siti (non sono elencate varianti nel file) |
cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.bam | file di allineamento in formato .bam (Binary Alignment Map) | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.bam.bai | file .bai (indice del file BAM) | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.bed | file .bed (Browser Extensible Data) | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.consensus.fa | genoma reference con rappresentazione di tutte le varianti | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.consensus.subs.fa | genoma reference con rappresentazione delle sole varianti di sostituzione | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.txt | riassunto della run di snippy | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.tab | tabella delle varianti in formato .tsv | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.csv | tabella delle varianti in formato .csv | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.filt.vcf | varianti filtrate da Freebayes | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.raw.vcf | varianti non filtrate da Freebayes | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.subs.vcf | tabella delle sole varianti di sostituzione in formato .vcf | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.gff | varianti in formato GFF3 | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.html | versione .html della tabella .tab delle varianti | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.vcf | output file di snippy (varianti identificate), in formato .vcf (header di riepilogo + tabella) | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.vcf.gz | output file .vcf compresso di snippy | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.vcf.gz.csi | indice bcftools del file .vcf.gz | cartella "result" |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID_coverage_plot.png | grafico della distribuzione del coverage | cartella "result" |
Per ulteriori informazioni sui files prodotti da snippy, i loro formati ed il loro contenuto, si invita a fare riferimento al manuale di snippy.
medaka
| File | Descrizione | Posizione |
|---|---|---|
| DSXXXXXXXX-DTXXXXXX_ID_medaka_REFID.fasta | file fasta dell'allineamento | cartella "result" |
Visualizzazione dei dati
Per valutare la qualità delle sequenze ottenute, l'allineamento delle reads sul reference può essere visualizzato usando software specifici (ad esempio Tablet, BioEdit e uGene) per leggere i file .bam e .bam.bai:
- Tablet (GNU/Linux, macOS, Windows): https://ics.hutton.ac.uk/tablet/download-tablet/;
- BioEdit (Windows): https://thalljiscience.github.io/;
- uGene (GNU/Linux, macOS, Windows): http://ugene.net/ugene/.