Pipeline Mapping for Segmented Virus
Introduzione
La pipeline Mapping for Segmented Virus permette di eseguire il mapping di frammenti di genoma di virus segmentati, confrontandoli con più reference. Questo permette di eseguire il mapping di ogni segmento sul reference appropriato. La pipeline produce, oltre ai singoli allineamenti, anche il multiFASTA completo.

Lancia la pipeline Mapping Virus Segmentati
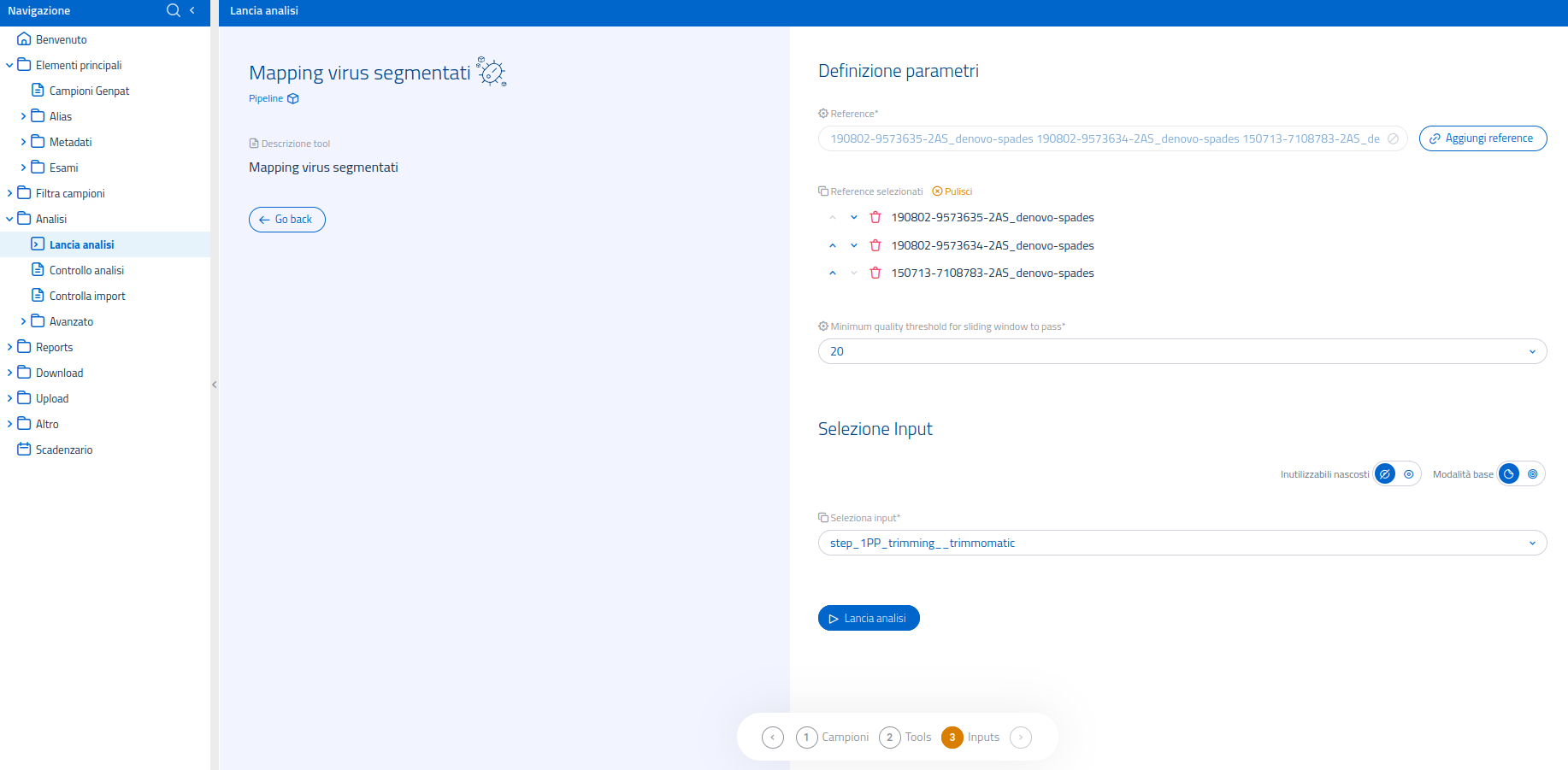
Nel sistema di lancio analisi, è possibile usare il filtro in alto per visualizzare esclusivamente le pipelines. Una volta selezionata la pipeline Mapping Virus Segmentati nella pagina dedicata al lancio di analisi, il sistema passerà ad un'interfaccia di conferma.
La pipeline Mapping Virus Segmentati esegue il mapping di tutti i segmenti di genoma, provenienti dai virus segmentati forniti, usando più references. I software usati saranno:
- Ivar (2AS_mapping__ivar) nei casi di Illumina short reads o IonTorrent;
- minimap2 (2AS_mapping__minimap2) per long reads Nanopore.
Gli input utilizzabili sono i risultati delle analisi di preprocessamento:
- reads deplete (step_1PP_hostdepl)
- downsampled reads (step_1PP_downsampling)
- reads trimmate (step_1PP_trimming)
- reads filtrate (step_1PP_filtering)
L'interfaccia per la selezione dell'input mette a disposizione la modalità di selezione input avanzata, per permettere l'utilizzo di input processati da metodi diversi, utilizzati a monte nel flusso di analisi.
Una volta lanciata la pipeline, la pagina genererà un link alla sezione Controllo analisi, per permettere di visualizzare lo stato del processo. L'utente verrà notificato dal sistema sia una volta lanciata con successo la pipeline, sia al termine dell'esecuzione.
Reference multipli
I reference multipli richiesti dalla pipeline devono essere forniti dall'utente tramite il sistema di selezione dedicato, la cui guida all'uso è consultabile nell'apposita sezione della Wiki sul lancio di analisi.
Risultati
I files prodotti dalla pipeline "Mapping Virus Segmentati" saranno gli stessi prodotti dalle analisi che la compongono, organizzati secondo la stessa gerarchia. Come riferimento, si rimanda quindi alla relativa sezione dell'analisi 2AS_mapping. A tali file, si aggiunge il multifasta dei segmenti di genoma mappati, le cui informazioni sono riportate nella seguente tabella:
| File | Descrizione | Posizione |
|---|---|---|
DSXXX-DTXXX_20XX.XX.XXXX.X_tool_aggregate.fasta |
multiFASTA con l'aggregazione delle sequenze consensus in un file unico | DSXXX-DTXXX_tool > result |
DSXXX-DTXXX_20XX.XX.XXXX.X_tool_reference.fasta |
FASTA con la sequenza consensus generata dal mapping su un singolo reference. I risultati di ogni mapping singolo sono salvati in una cartella identificata da un numero sequenziale addizionale, aggiunto al codice base della cartella | DSXXX-DTXXX[sequential-number]_tool > result |
DSXXX-DTXXX_20XX.XX.XXXX.X_tool_reference.bam e .bam.bai |
file bam e relativo indice generati dal mapping su un singolo reference. I risultati sono salvati come nel caso dei FASTA |
DSXXX-DTXXX[sequential-number]_tool > result |
| warnings.log | file di testo con avvertenze nel caso di numero di references non corretto | cartella "results" |